
Historical Perspective
The original concept of the pharmacophore was developed by Paul Ehrlich during the late 1800s. At that time, the understanding was that certain “chemical groups” or functions in a molecule were responsible for a biological effect, and molecules with similar effect had similar functions in common. The word pharmacophore was coined much later, by Schueler in his 1960 book Chemobiodynamics and Drug Design, and was defined as “a molecular framework that carries (phoros) the essential features responsible for a drug’s (pharmacon) biological activity.” The definition of a pharmacophore was therefore no longer concerned with “chemical groups” but “patterns of abstract features.” Since 1997, a pharmacophore has been defined by the International Union of Pure and Applied Chemistry as: A pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target and to trigger (or block) its biological response.
Introduction
A pharmacophore is an abstract description of molecular features which are necessary for molecular recognition of a ligand by a biological macromolecule. The pharmacophore should be considered as the largest common denominator of the molecular interaction features shared by a set of active molecules. Thus a pharmacophore does not represent a real molecule or a set of chemical groups, but is an abstract concept. Despite this clear definition, the term pharmacophore is often misused by many in medicinal chemistry to describe simple yet essential chemical functionalities in a molecule (such as guanidine or sulfonamides), or common chemical scaffolds (such as flavones or prostaglandins). Often the long definition is simplified as: “A pharmacophore is the pattern of features of a molecule that is responsible for a biological effect,” which captures the essential notion that a pharmacophore is built from features rather than defined chemical groups. A pharmacophore model explains how structurally diverse ligands can bind to a common receptor site. Furthermore, pharmacophore models can be used to identify through de novo design or virtual screening novel ligands that will bind to the same receptor.
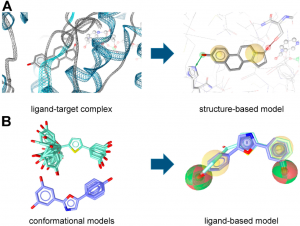
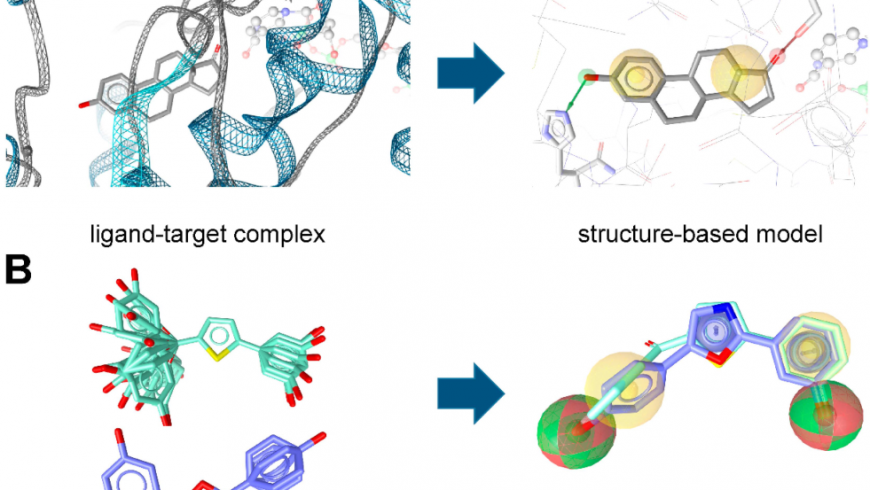
Fig1: (A) Structure- and (B) ligand-based pharmacophore model generation with LigandScout.
-
Based on the complex of equilin bound to 17β -HSD1 (PDB entry 1EQU), an initial pharmacophore model is created automatically (Sawicki et al., 1999);
-
Conformational models of known 17β -HSD1 ligands are used to align the compounds and extract pharmacophore features they share (Bey et al., 2008).
Pharmacophore Models
Pharmacophore models can be generated using two different approaches (Figure 1) depending on the input data employed for model construction. In the structure-based approach, the interaction pattern of a molecule and its targets are directly extracted from experimentally determined ligand-target complexes (Figure 1A). An important source for these complexes, e.g. derived from NMR-spectroscopy or X-ray crystallography, represents the Protein Data Bank (PDB, www.pdb.org). To date (access date 25 April, 2018), more than 1,44,840 macromolecular structures are stored in this online repository. However, not all of these structures were solved in a complex with a bound ligand, and in the case of induced fit, the binding of different ligands to an enzyme or receptor can lead to different interactions that are not covered by a single structure.
To address this limitation, some pharmacophore modeling programs, e.g. Discovery Studio and LigandScout, also provide tools to create pharmacophore models based exclusively on the topology of the binding site and in the absence of a ligand. In Discovery Studio, for example, the binding site can be defined manually by selecting residues within the desired cavity or by applying implemented binding site identification tools. Once the binding site is defined, the program automatically calculates pharmacophore features based on the residues lining the active site. This initial ensemble of pharmacophore features can then be adapted to construct the final hypothesis. In addition, structure-based pharmacophore models can also be generated with computationally derived ligand-target complexes. In the course of a docking run, known active compounds are fitted into the empty binding pocket of the target. These docked binding poses can then directly be employed to extract the interaction patterns. For further refinement of the initial docking poses, molecular dynamics (MD) simulations can be conducted prior to model generation.
In the course of ligand-based modeling, three-dimensional (3D) structures of two or more known active molecules are aligned and common pharmacophore features shared among these training set molecules are identified (Figure1 B). In a ligand-based approach, all of the common chemical features from the pharmacophore have to be presumed as essential, whereas in a structure-based approach, it can be considered whether a chemical feature of a molecule is directly involved in the ligand binding or not. Usually, datasets containing known active and inactive molecules are employed to assess the quality of the developed models. These datasets need to be designed carefully, because they largely influence the quality of the model and, accordingly, the success of the study. Only active molecules should be included, for which the direct interaction has been experimentally proven, e.g., by receptor binding or enzyme activity assays on isolated or recombinant proteins. Cell-based assays should be avoided in this context, because many factors other than interaction with the target can influence the results. Whenever no or only a limited number of known inactive molecules are available, so-called decoys (compounds with unknown biological activity but assumed to be inactive) might be employed.
These decoy-datasets need to be adapted for every target and should contain compounds with similar one-dimensional (1D) properties but different topologies compared to the known active molecules. These properties can include the number of hydrogen bond donors (HBDs), the number of hydrogen bond acceptors (HBAs), the number of non-polar atoms, molecular weight, logP, and the number of rotatable bonds. The Directory of Useful Decoys, Enhanced (DUD-E) provides a free service (http://dude.docking.org), where optimized decoys are generated based on the smiles codes of the uploaded active molecules. In general, a ratio of about 1:50 for the number of active molecules and decoys is recommended. This should reflect the prospective screening database, where usually only a few active molecules are also distributed among a vast amount of inactive molecules (Figure 2).
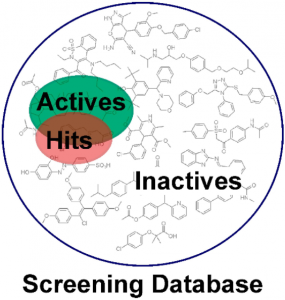 Fig2: Enrichment of active molecules in the virtual hit list. Usually, the majority of compounds in a screening database are inactive molecules, while a small pool of bioactive molecules is contained. Pharmacophore-based virtual screening can help to enrich active molecules in the hit list compared to a random selection of test compounds (Akram et al., 2015).
Fig2: Enrichment of active molecules in the virtual hit list. Usually, the majority of compounds in a screening database are inactive molecules, while a small pool of bioactive molecules is contained. Pharmacophore-based virtual screening can help to enrich active molecules in the hit list compared to a random selection of test compounds (Akram et al., 2015).
The preliminary models generated with both approaches need further improvement in the majority of cases to facilitate the recovery of the active molecules and concomitantly exclude the inactive compounds in the dataset from the hit list.
The aim of pharmacophore-based virtual screening (VS) is to enrich active molecules in a screening database in the virtual hit list (Figure 2). Multiple quality metrics are available that help to evaluate the quality of the developed pharmacophore model, for example the enrichment factor (the enrichment of active molecules compared to random selection), yield of actives (the percentage of active compounds in the virtual hit list), specificity (the ability to exclude inactive compounds) and sensitivity (the ability to identify active molecules), and the area under the curve of the Receiver Operating Characteristic plot (ROC-AUC).
The ultimate proof of a model’s quality and value, i.e., whether it is indeed capable of proposing novel active molecules, can, however, only be determined in a prospective experiment, as will be explained in more detail below. A workflow summarizing the individual steps of pharmacophore model generation and application is depicted in Figure 3.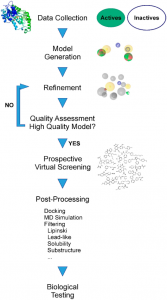
Fig 3: The different consecutive steps in pharmacophore model generation, refinement, and prospective application (Akram et al., 2015).
Pharmacophore Concepts in CADD
While the pharmacophore concept predates any form of electronic computer, it has nevertheless become an important tool in CADD. Every type of atom or group in a molecule that exhibits certain properties related to molecular recognition can be reduced to a pharmacophore feature. These molecular patterns can be labeled as hydrogen bond donors or acceptors, cationic, anionic, aromatic, or hydrophobic, and any possible combinations. Different molecules can be compared at the pharmacophore level; this usage is often described as “pharmacophore fingerprints.” When only a few pharmacophore features are considered in a 3D model the pharmacophore is sometimes described as a “query.”
Pharmacophore Fingerprint
While molecules are 3D entities, the pharmacophore representation reduces a molecule to a collection of features at the 2D or 3D level. A pharmacophore fingerprint is an extension of this concept, and typically annotates a molecule as a unique data string. All possible three-point or four-point sets of pharmacophore features (points) are enumerated for each ligand. The distance between the feature points is counted in bonds (for topological fingerprints), or by distance-binning when using 3D fingerprints (Fig 4).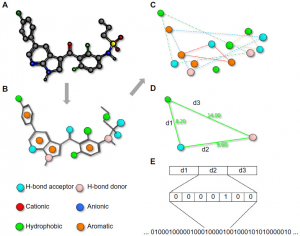 The resulting fingerprint is a string describing the frequency of every possible combination at predefined positions within the string. Several variants of pharmacophore fingerprints have been designed and are frequently used. Such a fingerprint can be used to analyze the similarity between molecules or among a library of molecules. Alternatively, a fingerprint model can be used to analyze the common elements of active ligands to identify the key contributing features to the biological function.
The resulting fingerprint is a string describing the frequency of every possible combination at predefined positions within the string. Several variants of pharmacophore fingerprints have been designed and are frequently used. Such a fingerprint can be used to analyze the similarity between molecules or among a library of molecules. Alternatively, a fingerprint model can be used to analyze the common elements of active ligands to identify the key contributing features to the biological function.
Fig 4: Pharmacophore fingerprints (Qing X et al., 2014).
Notes: A pharmacophore fingerprint is the representation of a small molecule ligand (A) annotated with molecular interaction features (B) into a string. Typically, every possible three- (or four-) point combination of molecular interaction features (C), with different distances between the features, calculated either through space or by the number of bond lengths (D), is calculated and the frequency of occurrence is stored in a string (E). Such strings are useful for the easy comparison of similarity between multiple molecules.
Pharmacophore modeling in virtual screening
Pharmacophore modeling is most often applied to virtual screening in order to identify molecules triggering the desired biological effect. For this purpose, researchers create a pharmacophore model (query) that most likely encodes the correct 3D organization of the required interaction pattern. Depending on how much is known about the particular protein target, different options are available to construct such a query (Fig 5).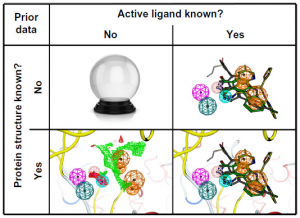
Fig 5: Four different situations for the pharmacophore search (Qing X et al., 2014)..
Notes: The figure shows the four different situations that may be encountered when starting a virtual screening. The situations include the absence of both the ligand and protein structure information, where except for divination, experimental screening is the only option. The second option is the presence of active ligands, but the protein structure is unknown, where pharmacophores can be used for ligand-based virtual screening. The best situation is when binding ligand and structural information is present. The most challenging option is when only a protein structure is available.
Applications of pharmacophores in ADME-tox
Poor ADME-tox is a major contributing factor to failures during drug development and clinical trials. It is, therefore, widely accepted that the ADME-tox properties should be profiled early during the drug discovery process, and pharmacophore modeling approaches are often used for such ADME-tox predictions. The pharmacophore models can be used to identify possible interactions of drugs with drug-metabolizing enzymes by matching the equivalent chemical groups of test molecules to those of drug molecules with a well-known ADME-tox profile. The enzymes of major importance for observed ADME-tox profile are the cytochrome P450s (CYP) that initiate drug breakdown.
Conclusion
Depending on the prior knowledge of the system, pharmacophores can be used to identify derivatives of compounds, change the scaffold to new compounds with a similar target, virtual screen for novel inhibitors, profile compounds for ADME-tox, investigate possible off-targets, or just complement other molecular methods. While there are limitations to the pharmacophore concept, multiple remedies are available at any time to counter them. Given this versatility, it is expected that pharmacophore modeling will maintain a dominant role in CADD for the foreseeable future, and any medicinal chemist should be aware of its benefits and possibilities.
References
-
Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A. and Schuster, D. (2015). Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases Molecules. Molecules. 20: 22799–22832; doi:10.3390/molecules201219880.
-
Qing, X.; Lee, X.Y.; De Raeymaecker, J.; Tame, J.; Zhang, K.; De Maeyer, M. and Voet, A. (2014). Pharmacophore modeling: advances, limitations, and current utility in drug discovery. Journal of Receptor, Lignad and Channel Research. 7: 81—92. https://doi.org/10.2147/JRLCR.S4684.
-
Sheng-Yong, Y. (2010). Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discovery Today. 15(11/12): https://doi.org/10.1016/j.drudis.2010.03.013.
Download PDF
Dr. Prabhakar Maurya1, Dr. Binita Angom2 and Dr. Luit M Barkalita3
1Medical Toxicology Division, Central Insecticides Board & Registration Committee, Haryana
2Dept. of Pharmacology, College of Veterinary Science & Animal Husbandry, Agartala, Tripura
3Dept. of Animal Biotechnology, College of Veterinary Science, AAU, Guwahati, Assam

