
Microbes comprise the overwhelming majority of life forms and are the most important organisms on Earth. It is stated that microbes run the world. Every process in the biosphere is touched by the microbes to transform the world around them. Most microorganisms live in communities or groups of more than one species. In these communities, they carry out their own functions and contribute to and depend on the activities of other microorganisms. Microbes have many properties similar to complex organisms like humans. There are numerous examples of essential services provided by microorganisms. Oxygen and the ozone layer, which are required for human life on Earth, were first generated entirely by microorganisms and today are maintained by photosynthetic bacteria along with plants. The global microbial diversity presents an enormous, largely untapped genetic and biological pool that could be exploited for the recovery of novel genes, metabolic pathways and valuable products. The microbial communities in and on the human body are essential to our health. Bacteria in the gastrointestinal tract protect us from invading pathogens; when the balance of gut microbial communities is compromised, many diseases, including inflammatory bowel disease, colon cancer and even obesity and diabetes, are thought to follow. These diseases are not incited by a single organism, but are mediated by community processes.
Viral infections are a major global health concern and new infectious diseases continue to emerge from year to year (Dong et al., 2008). Intensive globalization, climatic changes and viral evolution, among other factors, contributed for emergence of viruses and new viral diseases in the last decades. Animal pathogens viruses in particular, are considered to be a significant source of emerging human infections (Cleaveland et al., 2001). Identification and optimal characterization of novel viruses affecting both domestic and wild animal population is central to protecting both human and animal health. In this situation, it is crucial to apply powerful methods for broad-range detection and identification of the emerging viruses. According to a recent statistical estimate, there are at least 320,000 mammalian viruses that are yet to be discovered (Anthony et al., 2013). The conventional methods of virus detection has limitation to identify viruses that cannot be cultivated and are only characterized by molecular methods. Normally most of the diagnostic tests are designed to be virus-specific or aimed at a limited group of infectious agents. This makes them unsuitable for the detection of unexpected and/or completely new viruses as well as novel viral variants. Under these circumstances, viral metagenomics will be the most useful tool to sequence all viral genomes in a given sample without previous knowledge about their nature. Surveillance and monitoring of viral pathogens circulating in humans, animals and wildlife and the identification of emerging infectious diseases (EID) at an early stage is challenging. Many potential emerging viruses of concern might already be infecting humans or animals but await their detection by disease surveillance which can be possible through viral metagenomics (Temmam et al., 2014). Very recently, a novel surveillance strategy, using metagenomic analysis of toilet material from international air flights as a method for worldwide viral disease surveillance has been published (Hjelmso et al., 2019). The aim of the study was to design, implement and evaluate a method for viral analysis of airplane toilet waste enabling simultaneous detection and quantification of a wide range of human viral pathogens. Toilet waste from 19 international airplanes was analyzed for viral content using viral capture probes followed by high-throughput sequencing. Numerous human pathogens were detected including enteric and respiratory viruses. Several geographic trends were observed with samples originating from South Asia having significantly higher viral species richness as well as higher abundances of salivirus A, aichivirus A and enterovirus B, compared to samples originating from North Asia and North America (Fig.1).
Fig.1 Heatmap of the top 50 most abundant viruses and bacteriophages in the airplane toilet waste by metagenomics (Source: Hjelmsø et al., 2019)
Microorganisms are the source of most of the antibiotics that we use to manage infectious diseases. Microbes play a role in food production through fermentation, provide industrial enzymes and polymers, clean up toxic wastes, and protect humans and other animals and plants from diseases. Although microorganisms are often associated with diseases, pathogens represent only a tiny fraction of the total microbes on Earth. Unfortunately, scientists are able to grow in vitro only less than 1% of all microorganisms present in nature. This leaves more than 99% of the microbial diversity unexploited. One of the daunting challenges in studying microbial communities is gathering information from members that cannot be cultured under standard laboratory conditions. In some environments such as soil and ocean water, as few as 0.01–0.1% of the community members can be cultured. Metagenomics has revolutionized microbiology because it offers a window on an enormous and previously unknown world of microorganisms. It was first and foremost a method to learn about the contributions to the biosphere made by the microbial community members that could not be cultured. In this brief write-up, an introduction to metagenomics is given just to create awareness among the budding researchers.
The term metagenomics, the genomic analysis of a population of microorganisms, was coined by Handelsman and co-workers in 1998. In Greek, meta means “transcendent”. Metagenomics enable scientists to study all of the microbial genomes in a community as a whole, instead of studying genes and genome of individual microbes. Metagenomics has also been defined as the application of modern genomics techniques without the need for isolation and laboratory cultivation of individual microbial species. It is also called as the community genomics due to the isolation and analysis of total DNA from a population of microbial community without any prior cultivation (Chen and Petcher, 2005). Metagenomics basically refers to the study of the collective genomes of microbes in an environmental community and it makes it feasible to obtain information about entire microbial communities. For bacteria, 16s rRNA gene is universally used as a biomarker for microbial ecology studies due to very short (1542) nucleotide bases. There are nine hypervariable regions (V1–V9) that give sequence diversity for species identification. Hypervariable regions are conserved stretches enabling PCR amplification of target sequence using universal primers (Lu et al., 2000). But viruses, which lack such a universally conserved motif; hence viral metagenomics refers to the attempt to recover full and partial genomes of all viruses present in a sample (Nieuwenhuijse and Koopmans, 2017).
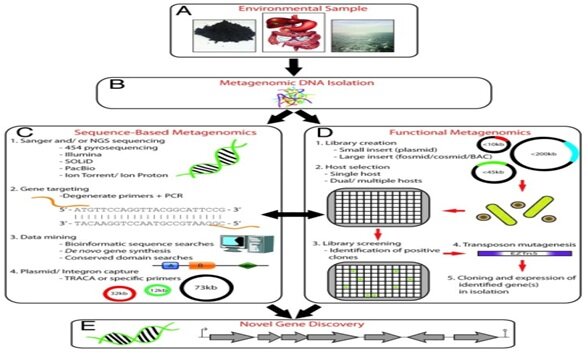
Metagenomics basically involves collection of appropriate samples from environmental source, extraction of DNA directly from an environmental sample and then studying the DNA sample. Metagenomics approaches are broadly divided into two categories. In the first approach, known as ‘sequence-driven metagenomics’, DNA extracted from the microbes in an environment of interest is sequenced and subjected to computational analysis. The metagenomic sequences are compared to sequences deposited in publicly available databases such as Genbank. Genes are then collected into groups of similar predicted function, and the distribution of various functions and types of proteins that conduct those functions can be assessed. In the second approach, ‘function-driven metagenomics’, the DNA extracted from the microbes in an environment is also captured and stored in a surrogate host, e.g. Escherichia coli, but instead of sequencing it, scientists screen the captured fragments of DNA or ‘clones’ for a certain function (Fig.2).
Fig.2. An overview of the main methods of metagenomics for novel gene discovery (Source: Culligan et al., 2013)
Metagenomics has emerged over the past two decades as an approach to elucidate a host of microbial communities inhabiting a specific niche with the goal of understanding their genetic diversity, population structure and ecological role. A number of new and novel molecules with significant functionalities and applications have been identified through this approach. The sustainable economic future of modern industrialized societies requires the development of novel molecules, enzymes, processes, products and applications. Metagenomics can also be applied to solve practical challenges in the field of medicine, agriculture, sustainability and ecology. Metagenomics promises to provide new molecules and novel enzymes with diverse functions and enhanced features compared to the enzymes from the culturable microorganisms. Besides the application of metagenomics for unlocking novel biocatalysts from nature, it also has found applications in fields as diverse as bioremediation, personalized medicine, xenobiotic metabolism, and so forth. Many microorganisms that have tight associations or symbiotic relationships with their hosts are difficult to culture away from the host; thus such symbiotic organisms were prime candidates for metagenomics. The communities of microbes that live symbiotically with human beings, known collectively as the ‘human microbiome’, are extraordinary in their complexity and influence on health. Metagenomic studies have revealed patterns that were invisible in studies relying on culture-based methods of study. For example, metagenomics and other culture-independent methods have demonstrated that each person carries a unique microbial community in his or her gastrointestinal tract; in fact, these communities have been called a ‘second fingerprint’ because they provide a personal signature for each of us. Unlike fingerprints, however, microbial communities are dynamic and change over our lifetimes because their composition is influenced by numerous factors. Metagenomic investigations on livestock and poultry species have shed light on the importance of the gut microbiome to feed efficiency, animal health, growth performance and productivity. Research on gut microbial diversity of different livestock and poultry species may help in identifying the probiotic communities that have both growth promoting and immunity boosting properties. The analysis of gut microbial diversity would help to find out the strains that are contributing to the adaptability of animals to the native conditions. There are limited metagenomic study in animal agriculture in India, the reasons behind may be poor infrastructure, cost of the study and availability of few skilled personnel. More studies are needed to be undertaken to explore the role of the microbial community in animal production and health.
Despite progress made in metagenomics approaches, there are many challenges in metogenomics and the important are – getting good quality sample from an environment, better DNA extraction techniques and analyses of the DNA sequences to study the functions of the proteins and metabolites generated by a community of microbes. Similarly, viral metagenomics suffers several pitfalls when considering new/highly divergent viral genomes. A critical challenge in viral metagenome assembly is the lack of a ubiquitous marker, analogous to bacterial 16S rRNA, to identify viral particles and estimate their diversity within ecological niches. Additionally, viral phylogeny based on sequences is impaired by extensive horizontal gene transfer and genome modularity within taxa, which is further complicated by the large numbers of viral particles within environmental samples. This makes it very difficult to find homologous sequences in reference database (Jorge et al., 2014).
Metagenomics provides a new lens for viewing the microbial world, particularly the huge uncultivable microbes and it has the potential to revolutionize understanding of the entire living world. Metagenomics will increasingly enhance our knowledge of microbial communities and can lead to major advancements in many areas, including human health, agriculture, energy production and environmental remediation. The emerging field of metagenomics opens the door to exploring the vast array of microbes unstudied to date and to investigating entire microbial communities. In the next decades, we expect that the top-down approach of metagenomics, the bottom up approach of classical microbiology and organism level genomics will merge. Metagenomic investigations have led to a better understanding of the microbiome and majority of the microbial genetic information generated is yet to be characterized and utilized.
References
Anthony, S.; Epstein, J.; Murray, K.; Navarrete-Macias, I.; Zambrana-Torrelio, C.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.; Islam, A. and Ali Khan, S. (2013). A strategy to estimate unknown viral diversity in mammals. Micro Biol., 4: 508-513.
Chen, K. and Pachter, L. (2005). Bioinformatics for whole genome shotgun sequencing of microbial communities. PLoS Comput. Biol., 1: 24.
Cleaveland, S.; Laurenson, M. and Taylor, L. (2001). Diseases of humans and their domestic mammals: Pathogen characteristics, host range and the risk of emergence. Biol. Sci., 356: 991-999.
Culligan, E.P.; Sleator, R.D.; Marchesi, J.R. and Hill, C.(2014). Metagenomics and novel gene discovery. Virulence, 5: 399–412.
Dong, J.; Olano, J.; McBride, J. and Walker, D. (2008). Emerging pathogens: challenges and successes of molecular diagnostics. J. Mol. Diagn., 10: 185-197.
Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J. and Goodman, R.M. (1998). Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol., 5: 245-249.
Hjelmso, M.H.; Mollerup, S.; Jensen, R.H.; Carlotta, P.; Lukjancenko, O.; Schultz, A.C.; Aarestrup, F.M. and Hansen, A.J. (2019). Metagenomic analysis of viruses in toilet waste from long distance flights – A new procedure for global infectious disease surveillance. Plos One, https://doi.org/10.1371/journal.pone.0210368.
Jorge, F.; Vazquez, C.; Rodrigo, G.L.V.; Brocal, M. and Pignatelli, M. (2014). Comparison of different assembly and annotation tools on analysis of simulated viral metagenomic communities in the gut. BMC Genomics, 15: 37.
Lu, J.J.; Perng, C.L.; Lee, S.Y. and Wan, C.C. (2000). Use of PCR with universal primers and restriction endonuclease digestions for detection and identification of common bacterial pathogens in cerebrospinal fluid. J. Clin. Microbiol., 38: 2076-2080.
Nieuwenhuijse, D. and Koopmans, M. (2017). Metagenomic sequencing for surveillance of food and waterborne viral diseases. Front. Microbiol., 8: 230.
Temmam, S.; Davoust, B.; Berenger, J.; Raoult, D. and Desnues, C. (2014). Viral metagenomics on animals as a tool for the detection of zoonoses prior to human infection. Int. J. Mol. Sci., 15: 10377-10397.
Dr. Dilip Kumar Sarma
Professor, Dept. of Microbiology,
College of Veterinary Science, A.A.U.
Khanapara, Guwahati-781022
Email: dksarma1956@gmail.com


